上皮樣血管平滑肌脂肪瘤(EAML)是血管平滑肌脂肪瘤的一種罕見變異,主要由上皮樣細胞組成,屬於血管周圍上皮樣細胞腫瘤(PEComa)家族。大多數EAML發生於腎臟,原發性肝EAML似乎比腎EAML少見。大多數PEComas是散發性的,但可能與結節性硬化症(TSC)有關——這是一種常染色體顯性遺傳性疾病,其特徵是TSC1或TSC2基因胚系突變。然而,此前曾報道過 5 名李法美尼綜合徵(Li-Fraumeni Syndrome,LFS)患者出現PEComas。LFS是一種遺傳性癌症易感綜合徵,由抑癌基因TP53的胚系突變引起。本文報告了一位患有肝EAML和胰腺癌的 49 歲女性患者。由於患者在 30 歲時確診患有雙側乳腺癌,因此研究人員進行了全面的基因分析,以確定與任何癌症易感綜合徵相關的基因變異。對患者血液樣本進行了全外顯子組測序(WES),發現了TP53(NM_000546.5):c.708C>A的雜合胚系變異;肝EAML和胰腺癌組織樣本的下一代測序(NGS)結果證實存在相同的TP53(NM_000546.5):c.708C>A胚系變異。結合患者的早發性乳腺癌病史,符合 2015 年版Chompret LFS診斷標準。關於LFS中存在PEComa的病例報告很少,據研究人員所知,這是LFS患者中首例肝EAML的報告。
背 景
李法美尼綜合徵(Li-Fraumeni Syndrome,LFS)是一種罕見的常染色體顯性癌症易感綜合徵,由Frederick P. Li和Joseph F. Fraumeni Jr於 1969 年首次描述。TP53胚系致病性變異於 1990 年被發現,並且仍是LFS的唯一已知原因。LFS的特點是罹患早發惡性腫瘤和多原發腫瘤的風險很高。肉瘤、乳腺癌、白血病、腦腫瘤和腎上腺皮質癌是最常見的惡性腫瘤,這些腫瘤統稱為「LFS core」腫瘤。
已經制定了幾套標準來識別可能攜帶TP53胚系突變的家族。經典的LFS定義包括先證者在 45 歲之前確診患有肉瘤、一級親屬在 45 歲之前罹患任何癌症,以及一級或二級親屬在 45 歲之前患有任何癌症或任何年齡罹患肉瘤。Birch和Eeles描述了更寬鬆的臨床定義,並用於描述「Li-Fraumeni樣綜合徵」,其中包括不屬於嚴格定義的特徵。最近,法國LFS工作組制定了用於識別TP53胚系突變患者的替代標準,稱為Chompret標準,該標準由Bougeard等人於 2015 年修訂。Chompret標準對於先證者的癌症類型和發病年齡有更多限制,但允許存在陰性家族史。其中包括提示LFS的四種臨床情況:家族中存在先證者(患有LFS腫瘤時年齡小於 46 歲[乳腺癌、軟組織肉瘤、骨肉瘤、中樞神經系統腫瘤、腎上腺皮質癌、白血病或支氣管肺泡肺癌])以及一名一級或二級親屬在 56 歲前患有LFS腫瘤或多原發腫瘤;多個原發性腫瘤(其中兩個屬於狹義LFS譜[narrow LFS spectrum],第一個腫瘤在發生在 46 歲之前);罕見癌症(腎上腺皮質癌或脈絡叢癌,無論家族史如何);早發性乳腺癌(31 歲之前)。
血管周圍上皮樣細胞腫瘤(PEComas)是一個腫瘤家族,包括血管平滑肌脂肪瘤(AML)、肺透明細胞癌、淋巴管平滑肌瘤病、原發性肺外透明細胞癌、鐮狀韌帶/圓韌帶和伴有血管周上皮樣細胞的腹盆腔肉瘤。AML由不同數量的黏液瘤、脂質和血管成分組成。AML的診斷需要通過免疫組化識別黑色素細胞標誌物的表達。
上皮樣血管平滑肌脂肪瘤(EAML)是AML的一種罕見變異,主要由上皮樣細胞組成。大多數EAML發生於腎臟,原發性肝EAML似乎比腎EAML少見。大多數PEComas是散發性的,但可能與結節性硬化症(TSC)相關,這是一種常染色體顯性遺傳病,由TSC1或TSC2基因胚系突變引起。然而,之前僅在 5 例LFS病例中報道過PEComas。本文報告一例LFS患者罹患肝EAML並伴有胰腺癌的病例。
病 例
患者女,49 歲,在體檢時通過腹部CT掃描偶然發現了直徑 3 cm的肝臟腫塊。患者在 30 歲時確診患有雙側乳腺癌,並接受了雙側改良乳房切除術。腹部核磁共振顯示肝臟和胰腺有兩個腫塊。右肝葉(第 6 段)的腫塊在T1加權圖像上顯示出低信號強度(圖1A),在T2加權圖像上呈中等信號強度(圖1B),難以與肝細胞癌鑑別。對肝臟腫塊行釓塞酸二鈉增強MRI,結果顯示動脈期圖像顯著增強,門脈期和延遲期上有可疑影像(圖1C-F)。研究人員在胰尾T2加權圖像上發現了一個高信號強度病變(圖2A)。對胰腺腫塊進行了釓塞酸二鈉增強MRI檢測,結果顯示未注射造影及前信號強度低,動脈期低增強,隨後在門靜脈期和延遲期出現延遲增強(圖2B)。
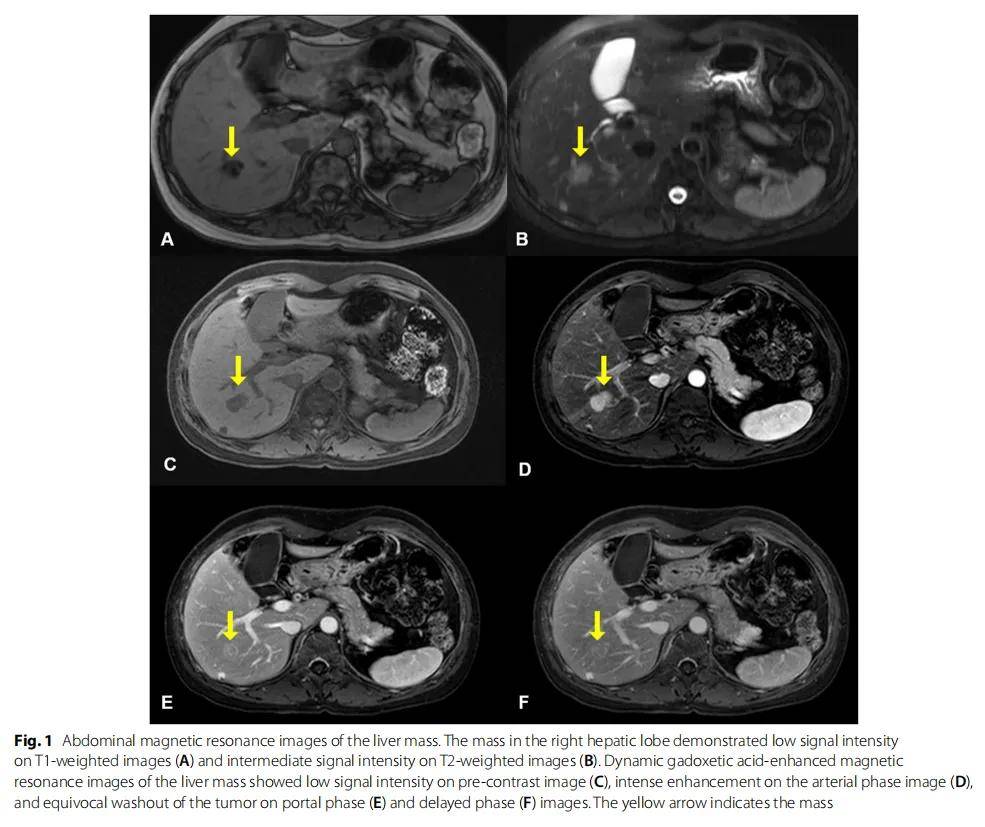
圖1 肝臟腫塊的腹部磁共振影像
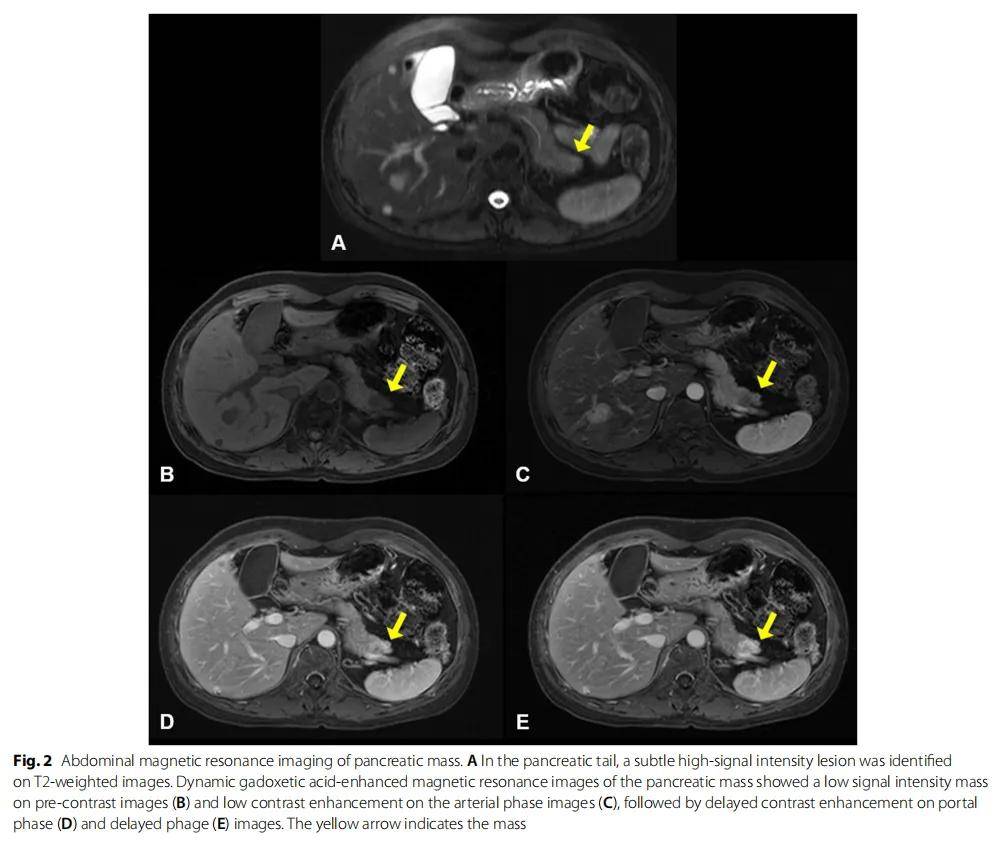
圖2 胰腺腫塊的腹部磁共振影像
隨後,患者接受了右肝切除術和遠端胰腺切除術。肝臟大體檢查發現毗鄰肝包膜處有一界限清晰的灰白色腫塊(2.2×2×1.1 cm)。顯微鏡下,腫塊與周圍的肝組織界限清楚(圖3A)。它由上皮樣細胞、脂肪細胞和薄壁血管組成(圖3B)。上皮樣細胞含有豐富的嗜酸性顆粒狀胞漿,細胞核大而圓,呈輕度異形性(圖3C)。未觀察到腫瘤壞死或有絲分裂。免疫組織化學顯示腫瘤細胞HMB45呈陽性,SMA局灶性陽性,但S-100、SOX10、肝細胞抗原和CK AE1/AE3呈陰性(圖3D、E)。腫瘤組織學特徵和免疫組化特徵與EAML一致。
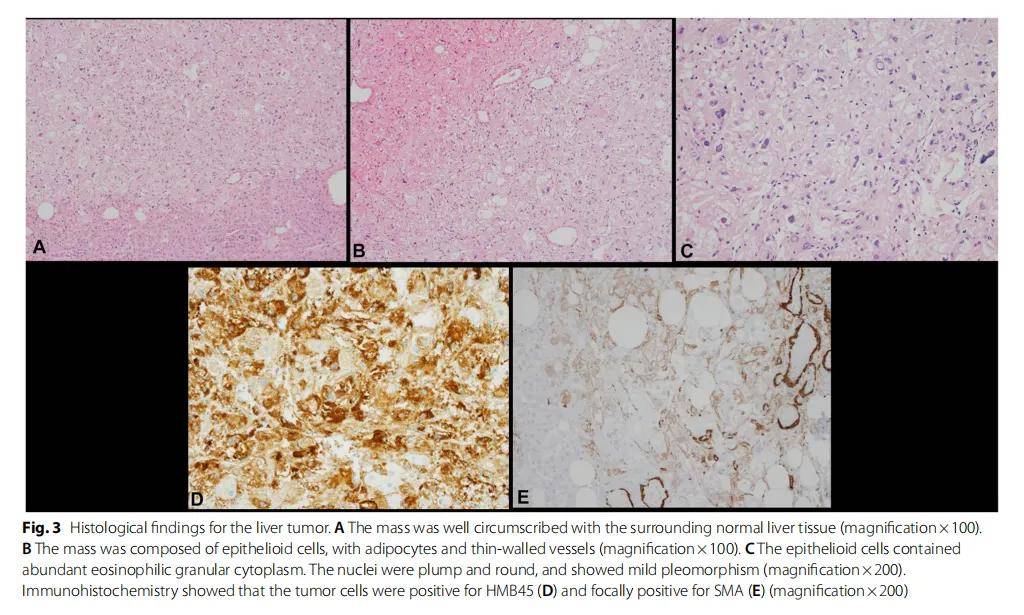
圖3 肝臟腫瘤的組織學結果
對遠端胰腺切除標本進行肉眼檢查,發現了一個邊界不清的灰白色實性病變,尺寸為 1.1×1×0.8 cm。組織病理學檢查發現胰腺中存在具有促纖維增生基質的非典型腺體(圖4A)。腺體由具有不規則核膜、突出核仁和管狀生長模式的細胞組成,與中分化胰腺腺癌相符(圖4B)。免疫組化顯示腫瘤細胞CK7陽性,CK20陰性(圖4C)。
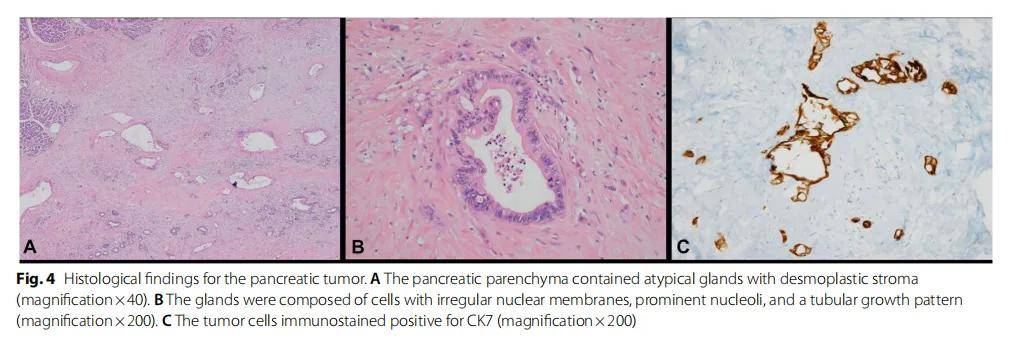
圖4 胰腺腫瘤的組織學結果
由於本例患者同時患有多個原發腫瘤,且有 30 歲時雙側早髮乳腺癌病史,存在遺傳性腫瘤易感綜合徵的可能性。患者確診乳腺癌時未進行基因檢測。本文詳細分析了患者家族病史,發現患者父親 45 歲時確診食管癌,妹妹 40 歲時確診乳腺癌。對患者的家系進行了調查,如圖5A所示。基於患者的癌症家族史,研究人員懷疑是家族性癌症易感綜合症。
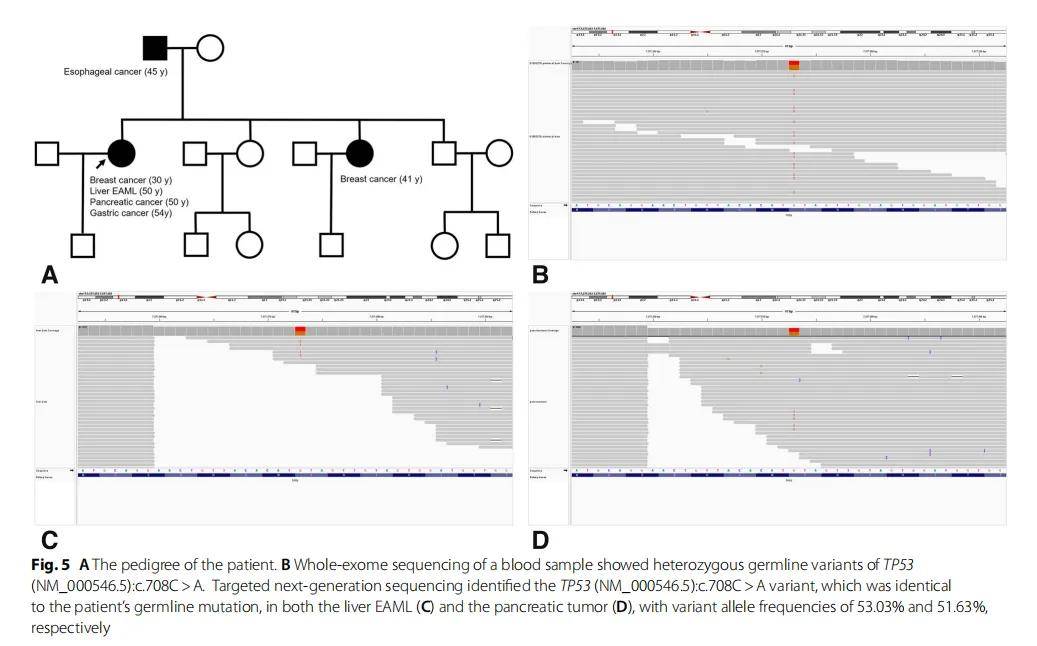
圖5
(A)患者家系;(B)血液全外顯子組測序結果;(C)肝臟腫瘤NGS結果;(D)胰腺腫瘤NGS結果
研究人員對患者的血液樣本進行了全外顯子組測序(WES),發現了TP53雜合胚系變異(NM_000546.5):c.708C>A(圖5B)。未發現有害的BRCA1/2突變。此外,研究人員還對肝EAML和胰腺腫瘤進行了NGS測序。NGS在肝EAML和胰腺癌中均檢出TP53(NM_000546.5):c.708C>A變異,變異等位基因頻率(VAF)分別為 53.05% 和 51.63%,這與患者的胚系相同突變(圖5C、D)。TP53變異的VAF也證實了TP53胚系突變的存在。再加上患者的早發性乳腺癌病史,符合Chompret 2015 年版LFS診斷標準。
TP53(NM_000546.5):c.708C>A變異尚未報道與LFS相關。在COSMIC資料庫中,有 19 個攜帶該變異的腫瘤樣本,但沒有一個是胚系突變。根據分子和臨床發現,ACMG將TP53(NM_000546.5):c.708C>A分類為「可能致病」,因為它是一種無義突變,導緻密碼子 236 處的酪氨酸殘基突變為終止密碼子(PVS1),在gnomAD外顯子組或基因組(PM2_supporting)中未找到。
術後 1 個月,患者接受5-氟尿嘧啶輔助化療 1 次,未見復發。然而 4 年後,通過食管胃十二指腸鏡檢查發現患者胃部有凹陷性病變(圖6A)。病變的組織病理學檢查診斷為原發性低黏附性胃癌(圖6B)。患者接受了胃大部切除術,最終病理TNM分類為pT1N1M0。未考慮進行胃癌輔助治療,3 年隨訪期間,患者仍然存活,沒有復發。
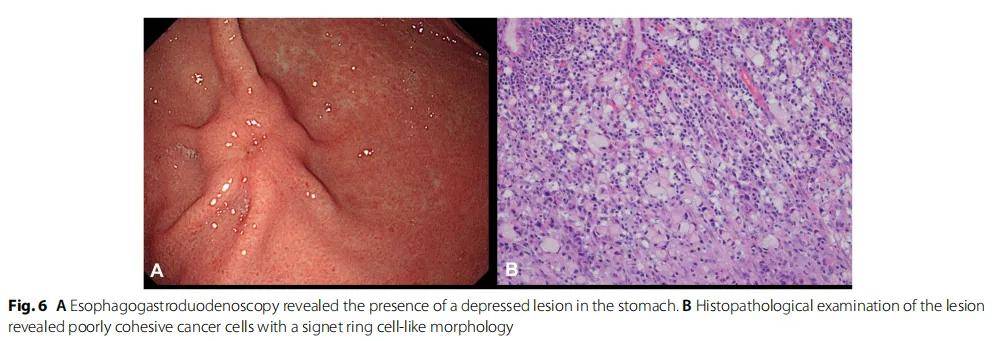
圖6
討 論
PEComas是一種罕見的間質腫瘤,惡性潛能不定。儘管大多數PEComas為散發病例,但PEComas的一個子集(~10%)可能與TSC(一種常染色體顯性多系統神經皮膚疾病)相關。散發性或遺傳性PEComas中最常見的基因變異是TSC1(~27%)或TSC2(~73%)基因功能缺失,導致mTOR信號傳導激活,這可能是一個治療靶點。此外,在一小部分PEComa患者(~23%)中報道了影響轉錄因子與IGHM增強子(TFE3)結合的基因重排。這具有重要的臨床意義,因為這些腫瘤可能對mTOR抑制劑沒有反應。最後,在 3 名子宮PEComa患者中發現了RAD51B易位。
EAML是AML的一種變體,其組織學以上皮樣為主,屬於PEComa家族。眾所周知,EAML更具侵襲性,並且與TSC的關係更密切。Lee等人報道肝EAML復發或轉移率為 9.3%,低於腎AML(30%)。據報道,約 6.2% 的肝EAML患者與TSC相關。
PEComa患者中也有TP53體系突變的報道。在一項系列研究中,有 8 例EAML(5 例腎臟,心臟、肝臟和子宮各一個),在一名肝EAML患者和兩名腎EAML患者中發現了TP53突變。此外,TP53突變可能與PEComa的惡性轉化有關。在一名患有腎AML的TSC患者中,從惡性上皮樣成分中發現了TP53錯義突變,但在典型的AML成分中沒有發現,這可能表明TP53在AML惡性轉化中發揮作用。最近,Akumulla等人對 31 例轉移性PEComas進行了全基因組分析,共檢出了 100 個基因變異。最常受影響的基因是TP53(45.2%)、TSC2(32.3%)、RB1(25.8%)、CDKN2A(19.3%)、TFE3(16.1%)、ATRX(9.6%)、TSC1(9.6%),以及CD36、FLCN、NF1和SMARCB1(各 6.4%)。在 16% 的腫瘤中還發現了TFE3重排。
迄今為止,已有 5 名LFS患者出現來自不同器官的PEComas報道。Neofytou等人報道了一名 24 歲患者同時患有肝臟和右腎的兩種原發性PEComas。Jasim等人描述了一名 26 歲的腎上腺EAML患者。Galera López等人報道了兩個患有LFS的兄弟姐妹同時診斷為肝臟PEComa。Butz等人報道了兩個患有LFS的兄姊同時診斷為肝臟PEComa。Butz等人報道了一名LFS患者大腿肌肉存在轉移性PEComa,該患者攜帶一種新的TP53胚系剪接突變。Caliskan等人報道了一名LFS患者出現子宮PEComa。在本文病例中,肝EAML與胰腺癌和LFS一起發生。通過對血液樣本進行全外顯子組測序和肝EAML的靶向測序,沒有發現既往報道的PEComa相關基因變異(包括TSC1、TSC2、TFE3和RAD51B)。
LFS是一種遺傳性癌症易感綜合徵,由TP53抑癌基因胚系突變引起。LFS相關腫瘤因其類型和患者終生患病率而異。與一般人群相比,兒童(0-15歲)罹患癌症的機率極高,癌症類型包括腎上腺皮質癌、脈絡叢癌、橫紋肌肉瘤或髓母細胞瘤;早期成人(16-50 歲)具有中風險,癌症形式包括乳腺癌、骨肉瘤、軟組織肉瘤、白血病、星形細胞瘤、膠質母細胞瘤、結直腸癌和肺癌;晚期成人(>50 歲)風險較低,癌症形式包括胰腺癌和前列腺癌。
隨著高通量基因檢測的廣泛應用,不符合臨床LFS基因檢測標準的、攜帶致病性/可能致病性(P/LP)TP53變異的患者個體正在被陸續識別,這些個體外顯率較低。因此,最近歐洲罕見遺傳腫瘤風險綜合徵網(ERN)建議通過使用「Li-Fraumeni譜系(Li-Fraumeni spectrum)」或「遺傳性TP53相關癌症(hTP53rc)綜合徵」等術語,將LFS的概念擴展到更廣泛的癌症易感綜合徵。
最近一項關於P/LP TP53胚系變異個體癌症發病率和模式的觀察性隊列研究表明,LFS患者的任何癌症發病率比一般人群高出近 24 倍。此外,近期有研究估計了普通人群中胚系P/LP TP53攜帶者的患病率,大約在 1:3,555–5,476 範圍內。TP53基因的檢測標準在過去三十年中已被廣泛討論,對於大多數腫瘤,如果根據家族和個人癌症史提示存在此類綜合徵,建議進行胚系檢測。
研究人員已確認胚系TP53(NM_000546.5):c.708C>A突變是與LFS相關肝EAML的新型致病性變異。雖然在COSMIC資料庫中已有報道,但均未獲證為胚系突變。由於該患者TP53基因存在突變,還符合 2015 年版Chompret LFS診斷標準,因此被診斷為LFS。
由於TP53是一種腫瘤抑制基因,通過調節細胞周期停滯、細胞凋亡和DNA修復來促進腫瘤發生,因此靶向TP53突變可能是一種合適的策略。最近,小分子p53激活劑PC14586獲得了FDA的快速通道認定,用於治療攜帶TP53 Y220C突變的晚期腫瘤。然而,目前對於LFS的治療尚無特殊建議。在治療LFS腫瘤期間,應儘量減少接觸放療和毒性化療,以避免原發性腫瘤的後續發展,特別是放療。因此,應優先進行手術或消融治療,儘可能避免放療,並根據最新指南使用非基因毒性化療。
本文描述了一名LFS患者罕見地發生肝EAML,該患者攜帶一種新型無義TP53突變。肝EAML是屬於PEComa家族的罕見腫瘤,與遺傳性癌症易感性疾病相關,特別是TSC。在極少數情況下,PEComas可能會與LFS一起出現。因此,對患者進行適當的PEComa診斷並對有風險的家庭成員進行篩查非常重要。
參考文獻:
Yang Y, Lee J, Woo CG, Lee OJ, Son SM. Epithelioid angiomyolipoma of the liver in a patient with Li-Fraumeni syndrome: a case report. Diagn Pathol. 2024 Jan 19;19(1):16. doi: 10.1186/s13000-023-01418-5. PMID: 38243242; PMCID: PMC10797712.