李法美尼综合征患者罹患肝上皮样血管平滑肌脂肪瘤,WES发现新型TP53胚系突变
上皮样血管平滑肌脂肪瘤(EAML)是血管平滑肌脂肪瘤的一种罕见变异,主要由上皮样细胞组成,属于血管周围上皮样细胞肿瘤(PEComa)家族。大多数EAML发生于肾脏,原发性肝EAML似乎比肾EAML少见。大多数PEComas是散发性的,但可能与结节性硬化症(TSC)有关——这是一种常染色体显性遗传性疾病,其特征是TSC1或TSC2基因胚系突变。然而,此前曾报道过 5 名李法美尼综合征(Li-Fraumeni Syndrome,LFS)患者出现PEComas。LFS是一种遗传性癌症易感综合征,由抑癌基因TP53的胚系突变引起。本文报告了一位患有肝EAML和胰腺癌的 49 岁女性患者。由于患者在 30 岁时确诊患有双侧乳腺癌,因此研究人员进行了全面的基因分析,以确定与任何癌症易感综合征相关的基因变异。对患者血液样本进行了全外显子组测序(WES),发现了TP53(NM_000546.5):c.708C>A的杂合胚系变异;肝EAML和胰腺癌组织样本的下一代测序(NGS)结果证实存在相同的TP53(NM_000546.5):c.708C>A胚系变异。结合患者的早发性乳腺癌病史,符合 2015 年版Chompret LFS诊断标准。关于LFS中存在PEComa的病例报告很少,据研究人员所知,这是LFS患者中首例肝EAML的报告。
背 景
李法美尼综合征(Li-Fraumeni Syndrome,LFS)是一种罕见的常染色体显性癌症易感综合征,由Frederick P. Li和Joseph F. Fraumeni Jr于 1969 年首次描述。TP53胚系致病性变异于 1990 年被发现,并且仍是LFS的唯一已知原因。LFS的特点是罹患早发恶性肿瘤和多原发肿瘤的风险很高。肉瘤、乳腺癌、白血病、脑肿瘤和肾上腺皮质癌是最常见的恶性肿瘤,这些肿瘤统称为“LFS core”肿瘤。
已经制定了几套标准来识别可能携带TP53胚系突变的家族。经典的LFS定义包括先证者在 45 岁之前确诊患有肉瘤、一级亲属在 45 岁之前罹患任何癌症,以及一级或二级亲属在 45 岁之前患有任何癌症或任何年龄罹患肉瘤。Birch和Eeles描述了更宽松的临床定义,并用于描述“Li-Fraumeni样综合征”,其中包括不属于严格定义的特征。最近,法国LFS工作组制定了用于识别TP53胚系突变患者的替代标准,称为Chompret标准,该标准由Bougeard等人于 2015 年修订。Chompret标准对于先证者的癌症类型和发病年龄有更多限制,但允许存在阴性家族史。其中包括提示LFS的四种临床情况:家族中存在先证者(患有LFS肿瘤时年龄小于 46 岁[乳腺癌、软组织肉瘤、骨肉瘤、中枢神经系统肿瘤、肾上腺皮质癌、白血病或支气管肺泡肺癌])以及一名一级或二级亲属在 56 岁前患有LFS肿瘤或多原发肿瘤;多个原发性肿瘤(其中两个属于狭义LFS谱[narrow LFS spectrum],第一个肿瘤在发生在 46 岁之前);罕见癌症(肾上腺皮质癌或脉络丛癌,无论家族史如何);早发性乳腺癌(31 岁之前)。
血管周围上皮样细胞肿瘤(PEComas)是一个肿瘤家族,包括血管平滑肌脂肪瘤(AML)、肺透明细胞癌、淋巴管平滑肌瘤病、原发性肺外透明细胞癌、镰状韧带/圆韧带和伴有血管周上皮样细胞的腹盆腔肉瘤。AML由不同数量的黏液瘤、脂质和血管成分组成。AML的诊断需要通过免疫组化识别黑色素细胞标志物的表达。
上皮样血管平滑肌脂肪瘤(EAML)是AML的一种罕见变异,主要由上皮样细胞组成。大多数EAML发生于肾脏,原发性肝EAML似乎比肾EAML少见。大多数PEComas是散发性的,但可能与结节性硬化症(TSC)相关,这是一种常染色体显性遗传病,由TSC1或TSC2基因胚系突变引起。然而,之前仅在 5 例LFS病例中报道过PEComas。本文报告一例LFS患者罹患肝EAML并伴有胰腺癌的病例。
病 例
患者女,49 岁,在体检时通过腹部CT扫描偶然发现了直径 3 cm的肝脏肿块。患者在 30 岁时确诊患有双侧乳腺癌,并接受了双侧改良乳房切除术。腹部核磁共振显示肝脏和胰腺有两个肿块。右肝叶(第 6 段)的肿块在T1加权图像上显示出低信号强度(图1A),在T2加权图像上呈中等信号强度(图1B),难以与肝细胞癌鉴别。对肝脏肿块行钆塞酸二钠增强MRI,结果显示动脉期图像显著增强,门脉期和延迟期上有可疑影像(图1C-F)。研究人员在胰尾T2加权图像上发现了一个高信号强度病变(图2A)。对胰腺肿块进行了钆塞酸二钠增强MRI检测,结果显示未注射造影及前信号强度低,动脉期低增强,随后在门静脉期和延迟期出现延迟增强(图2B)。
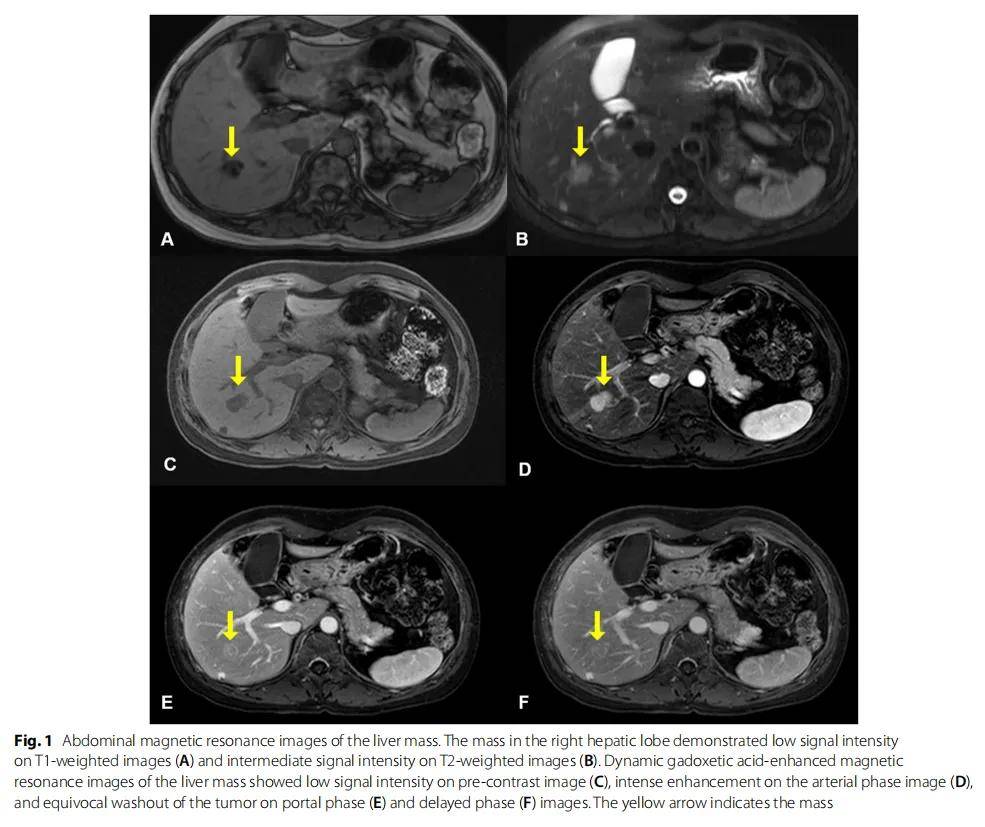
图1 肝脏肿块的腹部磁共振影像
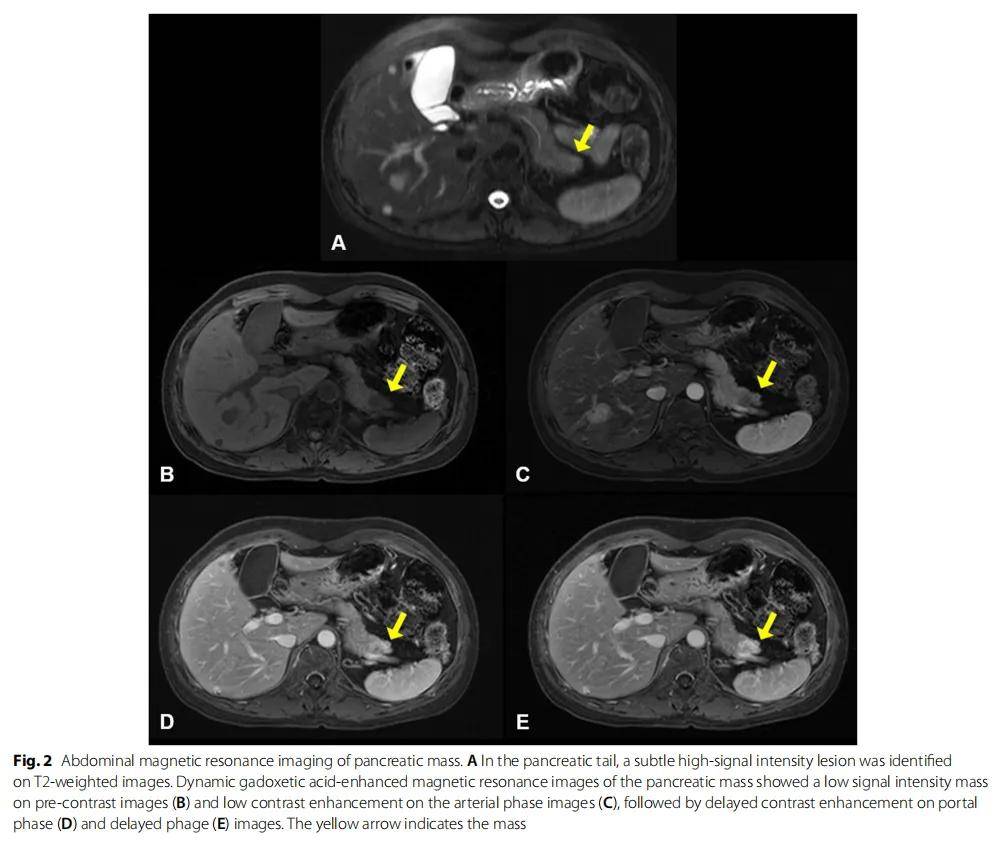
图2 胰腺肿块的腹部磁共振影像
随后,患者接受了右肝切除术和远端胰腺切除术。肝脏大体检查发现毗邻肝包膜处有一界限清晰的灰白色肿块(2.2×2×1.1 cm)。显微镜下,肿块与周围的肝组织界限清楚(图3A)。它由上皮样细胞、脂肪细胞和薄壁血管组成(图3B)。上皮样细胞含有丰富的嗜酸性颗粒状胞浆,细胞核大而圆,呈轻度异形性(图3C)。未观察到肿瘤坏死或有丝分裂。免疫组织化学显示肿瘤细胞HMB45呈阳性,SMA局灶性阳性,但S-100、SOX10、肝细胞抗原和CK AE1/AE3呈阴性(图3D、E)。肿瘤组织学特征和免疫组化特征与EAML一致。
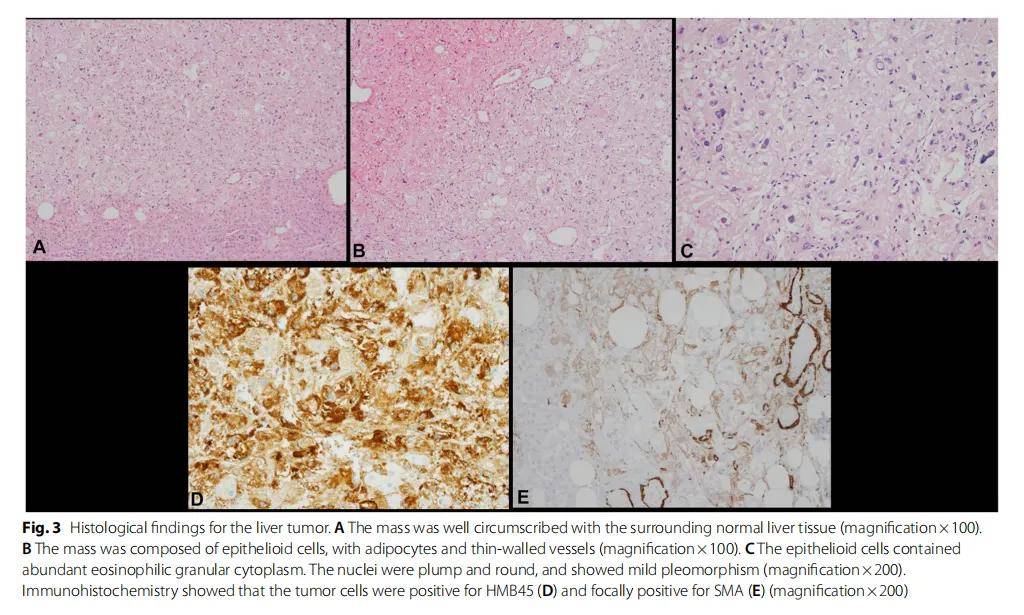
图3 肝脏肿瘤的组织学结果
对远端胰腺切除标本进行肉眼检查,发现了一个边界不清的灰白色实性病变,尺寸为 1.1×1×0.8 cm。组织病理学检查发现胰腺中存在具有促纤维增生基质的非典型腺体(图4A)。腺体由具有不规则核膜、突出核仁和管状生长模式的细胞组成,与中分化胰腺腺癌相符(图4B)。免疫组化显示肿瘤细胞CK7阳性,CK20阴性(图4C)。
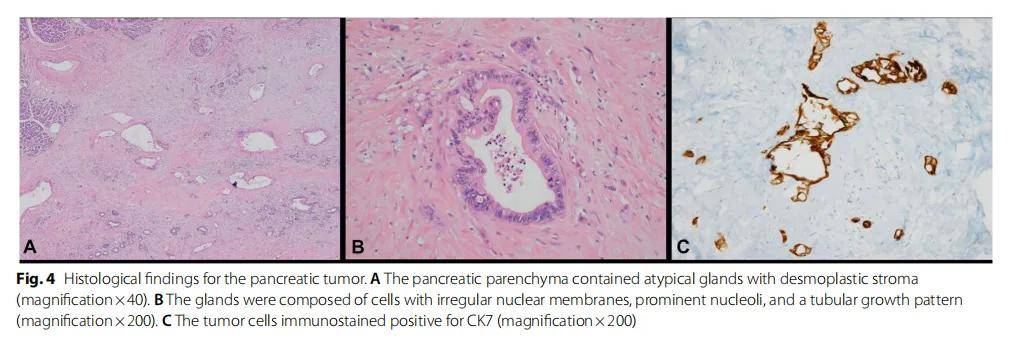
图4 胰腺肿瘤的组织学结果
由于本例患者同时患有多个原发肿瘤,且有 30 岁时双侧早发乳腺癌病史,存在遗传性肿瘤易感综合征的可能性。患者确诊乳腺癌时未进行基因检测。本文详细分析了患者家族病史,发现患者父亲 45 岁时确诊食管癌,妹妹 40 岁时确诊乳腺癌。对患者的家系进行了调查,如图5A所示。基于患者的癌症家族史,研究人员怀疑是家族性癌症易感综合症。
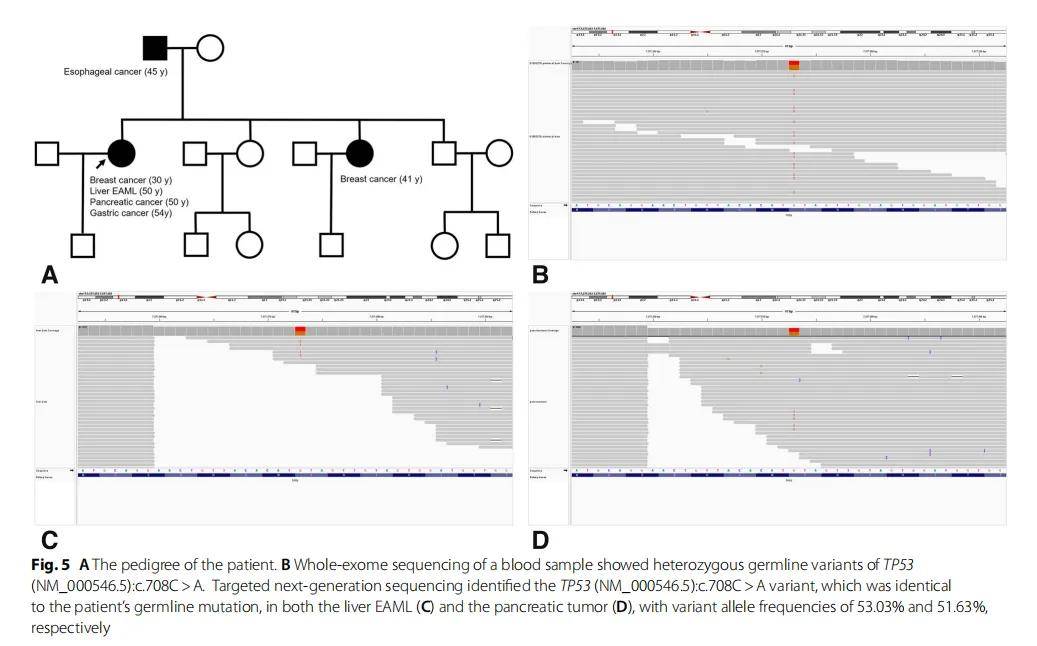
图5
(A)患者家系;(B)血液全外显子组测序结果;(C)肝脏肿瘤NGS结果;(D)胰腺肿瘤NGS结果
研究人员对患者的血液样本进行了全外显子组测序(WES),发现了TP53杂合胚系变异(NM_000546.5):c.708C>A(图5B)。未发现有害的BRCA1/2突变。此外,研究人员还对肝EAML和胰腺肿瘤进行了NGS测序。NGS在肝EAML和胰腺癌中均检出TP53(NM_000546.5):c.708C>A变异,变异等位基因频率(VAF)分别为 53.05% 和 51.63%,这与患者的胚系相同突变(图5C、D)。TP53变异的VAF也证实了TP53胚系突变的存在。再加上患者的早发性乳腺癌病史,符合Chompret 2015 年版LFS诊断标准。
TP53(NM_000546.5):c.708C>A变异尚未报道与LFS相关。在COSMIC数据库中,有 19 个携带该变异的肿瘤样本,但没有一个是胚系突变。根据分子和临床发现,ACMG将TP53(NM_000546.5):c.708C>A分类为“可能致病”,因为它是一种无义突变,导致密码子 236 处的酪氨酸残基突变为终止密码子(PVS1),在gnomAD外显子组或基因组(PM2_supporting)中未找到。
术后 1 个月,患者接受5-氟尿嘧啶辅助化疗 1 次,未见复发。然而 4 年后,通过食管胃十二指肠镜检查发现患者胃部有凹陷性病变(图6A)。病变的组织病理学检查诊断为原发性低黏附性胃癌(图6B)。患者接受了胃大部切除术,最终病理TNM分类为pT1N1M0。未考虑进行胃癌辅助治疗,3 年随访期间,患者仍然存活,没有复发。
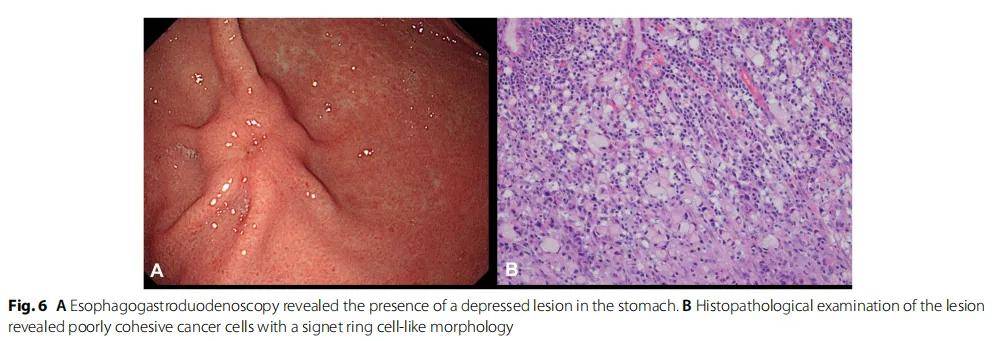
图6
讨 论
PEComas是一种罕见的间质肿瘤,恶性潜能不定。尽管大多数PEComas为散发病例,但PEComas的一个子集(~10%)可能与TSC(一种常染色体显性多系统神经皮肤疾病)相关。散发性或遗传性PEComas中最常见的基因变异是TSC1(~27%)或TSC2(~73%)基因功能缺失,导致mTOR信号传导激活,这可能是一个治疗靶点。此外,在一小部分PEComa患者(~23%)中报道了影响转录因子与IGHM增强子(TFE3)结合的基因重排。这具有重要的临床意义,因为这些肿瘤可能对mTOR抑制剂没有反应。最后,在 3 名子宫PEComa患者中发现了RAD51B易位。
EAML是AML的一种变体,其组织学以上皮样为主,属于PEComa家族。众所周知,EAML更具侵袭性,并且与TSC的关系更密切。Lee等人报道肝EAML复发或转移率为 9.3%,低于肾AML(30%)。据报道,约 6.2% 的肝EAML患者与TSC相关。
PEComa患者中也有TP53体系突变的报道。在一项系列研究中,有 8 例EAML(5 例肾脏,心脏、肝脏和子宫各一个),在一名肝EAML患者和两名肾EAML患者中发现了TP53突变。此外,TP53突变可能与PEComa的恶性转化有关。在一名患有肾AML的TSC患者中,从恶性上皮样成分中发现了TP53错义突变,但在典型的AML成分中没有发现,这可能表明TP53在AML恶性转化中发挥作用。最近,Akumulla等人对 31 例转移性PEComas进行了全基因组分析,共检出了 100 个基因变异。最常受影响的基因是TP53(45.2%)、TSC2(32.3%)、RB1(25.8%)、CDKN2A(19.3%)、TFE3(16.1%)、ATRX(9.6%)、TSC1(9.6%),以及CD36、FLCN、NF1和SMARCB1(各 6.4%)。在 16% 的肿瘤中还发现了TFE3重排。
迄今为止,已有 5 名LFS患者出现来自不同器官的PEComas报道。Neofytou等人报道了一名 24 岁患者同时患有肝脏和右肾的两种原发性PEComas。Jasim等人描述了一名 26 岁的肾上腺EAML患者。Galera López等人报道了两个患有LFS的兄弟姐妹同时诊断为肝脏PEComa。Butz等人报道了两个患有LFS的兄姊同时诊断为肝脏PEComa。Butz等人报道了一名LFS患者大腿肌肉存在转移性PEComa,该患者携带一种新的TP53胚系剪接突变。Caliskan等人报道了一名LFS患者出现子宫PEComa。在本文病例中,肝EAML与胰腺癌和LFS一起发生。通过对血液样本进行全外显子组测序和肝EAML的靶向测序,没有发现既往报道的PEComa相关基因变异(包括TSC1、TSC2、TFE3和RAD51B)。
LFS是一种遗传性癌症易感综合征,由TP53抑癌基因胚系突变引起。LFS相关肿瘤因其类型和患者终生患病率而异。与一般人群相比,儿童(0-15岁)罹患癌症的概率极高,癌症类型包括肾上腺皮质癌、脉络丛癌、横纹肌肉瘤或髓母细胞瘤;早期成人(16-50 岁)具有中风险,癌症形式包括乳腺癌、骨肉瘤、软组织肉瘤、白血病、星形细胞瘤、胶质母细胞瘤、结直肠癌和肺癌;晚期成人(>50 岁)风险较低,癌症形式包括胰腺癌和前列腺癌。
随着高通量基因检测的广泛应用,不符合临床LFS基因检测标准的、携带致病性/可能致病性(P/LP)TP53变异的患者个体正在被陆续识别,这些个体外显率较低。因此,最近欧洲罕见遗传肿瘤风险综合征网(ERN)建议通过使用“Li-Fraumeni谱系(Li-Fraumeni spectrum)”或“遗传性TP53相关癌症(hTP53rc)综合征”等术语,将LFS的概念扩展到更广泛的癌症易感综合征。
最近一项关于P/LP TP53胚系变异个体癌症发病率和模式的观察性队列研究表明,LFS患者的任何癌症发病率比一般人群高出近 24 倍。此外,近期有研究估计了普通人群中胚系P/LP TP53携带者的患病率,大约在 1:3,555–5,476 范围内。TP53基因的检测标准在过去三十年中已被广泛讨论,对于大多数肿瘤,如果根据家族和个人癌症史提示存在此类综合征,建议进行胚系检测。
研究人员已确认胚系TP53(NM_000546.5):c.708C>A突变是与LFS相关肝EAML的新型致病性变异。虽然在COSMIC数据库中已有报道,但均未获证为胚系突变。由于该患者TP53基因存在突变,还符合 2015 年版Chompret LFS诊断标准,因此被诊断为LFS。
由于TP53是一种肿瘤抑制基因,通过调节细胞周期停滞、细胞凋亡和DNA修复来促进肿瘤发生,因此靶向TP53突变可能是一种合适的策略。最近,小分子p53激活剂PC14586获得了FDA的快速通道认定,用于治疗携带TP53 Y220C突变的晚期肿瘤。然而,目前对于LFS的治疗尚无特殊建议。在治疗LFS肿瘤期间,应尽量减少接触放疗和毒性化疗,以避免原发性肿瘤的后续发展,特别是放疗。因此,应优先进行手术或消融治疗,尽可能避免放疗,并根据最新指南使用非基因毒性化疗。
本文描述了一名LFS患者罕见地发生肝EAML,该患者携带一种新型无义TP53突变。肝EAML是属于PEComa家族的罕见肿瘤,与遗传性癌症易感性疾病相关,特别是TSC。在极少数情况下,PEComas可能会与LFS一起出现。因此,对患者进行适当的PEComa诊断并对有风险的家庭成员进行筛查非常重要。
参考文献:
Yang Y, Lee J, Woo CG, Lee OJ, Son SM. Epithelioid angiomyolipoma of the liver in a patient with Li-Fraumeni syndrome: a case report. Diagn Pathol. 2024 Jan 19;19(1):16. doi: 10.1186/s13000-023-01418-5. PMID: 38243242; PMCID: PMC10797712.